Page Not Found
Page not found. Your pixels are in another canvas.
A list of all the posts and pages found on the site. For you robots out there is an XML version available for digesting as well.
Page not found. Your pixels are in another canvas.
About me
This is a page not in th emain menu
Uploaded:
Uploaded:


Published: AEM
we provide an extensive review, focusing on the application of MD simulations to decode the electrochemical processes in LIBs. To our knowledge, this is the first article to provide an exhaustive review of the electrochemical processes within electrodes, electrolytes, and electrode-electrolyte interfaces LIBs, exclusively from the perspective of pure MD simulations. Advancing further, this article elucidates the state-of-the-art advancements in MD simulation applications for a diverse range of electrochemical processes across different LIB components. Building on this foundation, our review then delves into the following three critical dimensions:
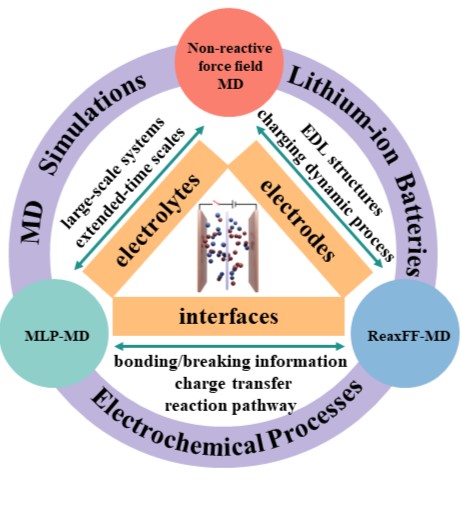
Citation: Xi Tan, Ming Chen, Jinkai Zhang, Shiqi Li, Huajie Zhang, Long Yang, Tian Sun, Xin Qian*, Guang Feng*, Decoding electrochemical processes of lithium-ion batteries by classical molecular dynamics simulations, *Advanced Energy Materials*, 2024, 14, 2400564.
Paper Link: https://onlinelibrary.wiley.com/doi/10.1002/aenm.202400564
Published: JCP
This review comprehensively examines molecular dynamics simulations of electrochemical interfaces, specifically addressing the utilization and implementation of constant potential methods within this domain. The principal objective of this paper is to elucidate the practical utility and potential implications of employing constant potential methods in the study of electrochemical interfaces, thereby providing an in-depth insight into the intricate interactions and dynamics at the molecular level.
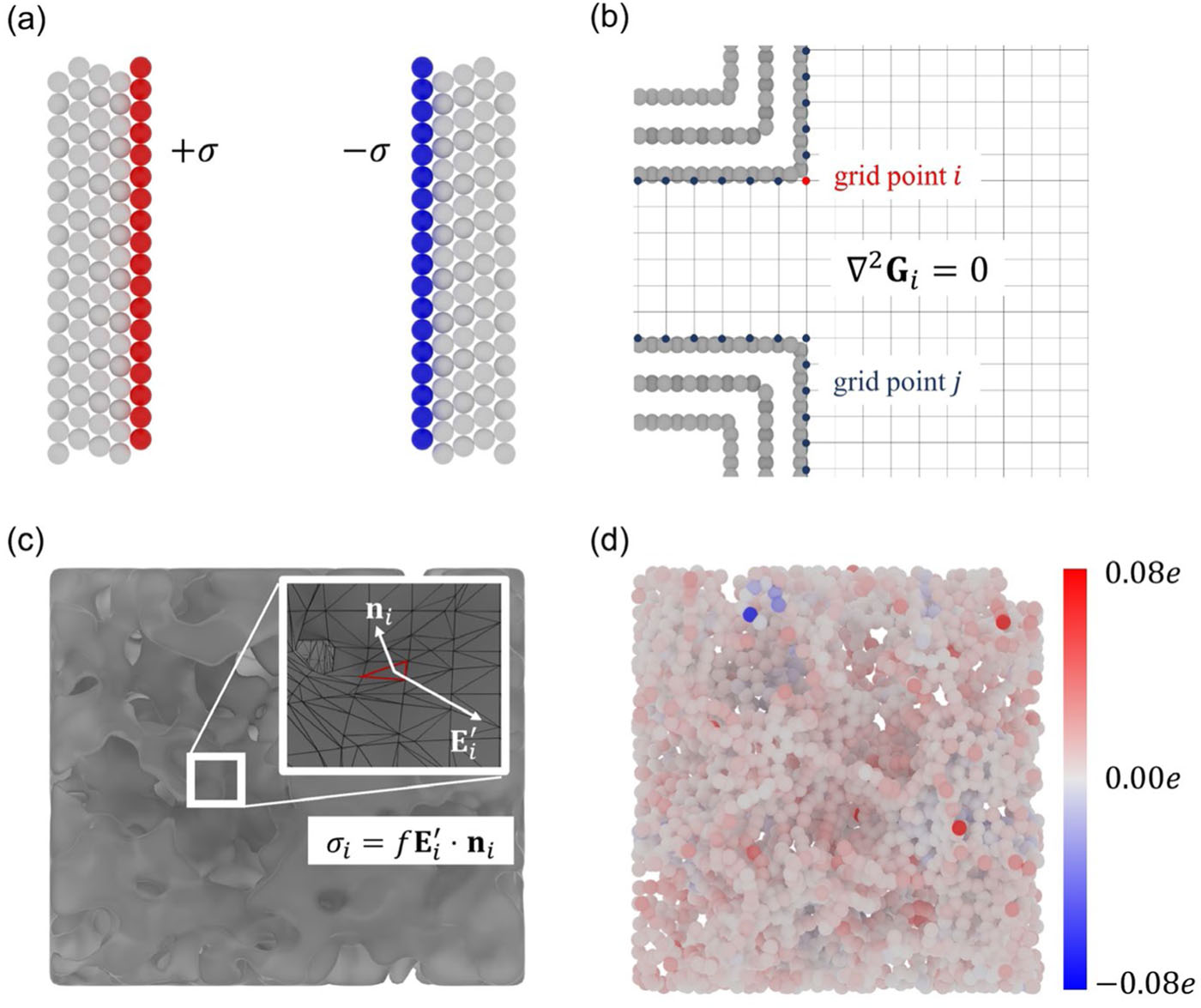
Citation: Liang Zeng, Jiaxing Peng, Jinkai Zhang, Xi Tan, Xiangyu Ji, Shiqi Li, Guang Feng*, Molecular dynamics simulations of electrochemical interfaces, The Journal of Chemical Physics, 2023, 159(9), 091001.
Paper Link: https://pubs.aip.org/aip/jcp/article/159/9/091001/2909729
Published: JEC
Molecular simulation can accurately depict the atomic-level interface structure of the electric double layer and its dynamic formation process, becoming an essential tool in exploring related fields. In the molecular simulation of the electric double layer, describing electrode polarization is crucial. Constant charge method (CCM) and constant potential method (CPM) are two important approaches. The constant charge method achieves electrode polarization by uniformly distributing charge on the electrode surface, which makes it easier to use and requires fewer computational resources, thus being widely utilized in relevant research. However, the constant charge method does not ensure the physical essence of the electrode being at a constant potential, making it theoretically less accurate. In contrast, the constant potential method maintains the electrode at a constant potential through dynamic changes in electrode atomic charges, aligning more closely with real conditions and yielding more accurate results. However, due to the real-time accurate solution of electrode atomic charges, the constant potential method requires nearly an order of magnitude more computational resources.

Citation: Liang Zeng, Xi Tan, Xiangyu Ji, Shiqi Li, Jinkai Zhang, Jiaxing Peng, Sheng Bi, Guang Feng*. Constant charge method or constant potential method: Which is better for molecular modeling of electrical double layers? Journal of Energy Chemistry, 2024, 94, 54.
Paper Link: https://www.sciencedirect.com/science/article/pii/S2095495624001694
Citation: xxx
Paper Link: waiting
Undergraduate course, University 1, Department, 2014
This is a description of a teaching experience. You can use markdown like any other post.
Workshop, University 1, Department, 2015
This is a description of a teaching experience. You can use markdown like any other post.