NEP Machine Learning Potential Training for NiHAB Metal-organic Framework
Published:
Quickly get started with MLP training-Solid materials
1. Training process
1) To ensure precise training structures, we executed simulations using the VASP 6.4.1 software on two AIMD-ML-NPT samples at temperatures of 298 K and a range of 200K to 500K. These simulations were conducted with a step size of 0.50 fs under a pressure of 1 bar, completing 100,000 steps. Detailed AIMD settings are available in the appendix.
2) From the 2 AIMD-ML trajectories, we uniformly extracted 50 structures, designating them as our training set.
3)Perform high-precision DFT static calculations on all the above structures.
4) Subsequently, we utilized GPUMD to train the NEP. Mirroring our earlier approach with room temperature water, we employed the active learning method. Utilizing the aforementioned 100 AIMD-ML samples from the NiHAB structure, the initial NEP was trained. We then executed NEP-MD active learning iterations at 298K and 1atm. Notably, our efforts resulted in the attainment of a first-principles-accurate MLP after only one iteration, involving just 7 additional structures (for accuracy validation, refer to Section 2). Comprehensive details on the NEP training parameters are delineated in the appendix.
In conclusion, it was a remarkable discovery that a high-precision MLP for the 2D MOF-NiHAB was achievable with merely 107 structures. This underscores the formidable learning capacity of the NEP, coupled with its distinctive resource efficiency.
2.NEP Machine Learning Potential accuracy verification
To ensure the credibility and precision of our trained machine learning potential, it is imperative to validate properties such as the Radial Distribution Function (RDF) and density. For a more nuanced assessment of our NEP’s accuracy, we conducted an AIMD without the on-the-fly acceleration, providing a benchmark for more accurate comparisons. Our analysis encompassed simultaneous comparisons among AIMD, AIMD_ML (using the on-the-fly method), and NEP-MD. The results are delineated below:
1) Loss train and test
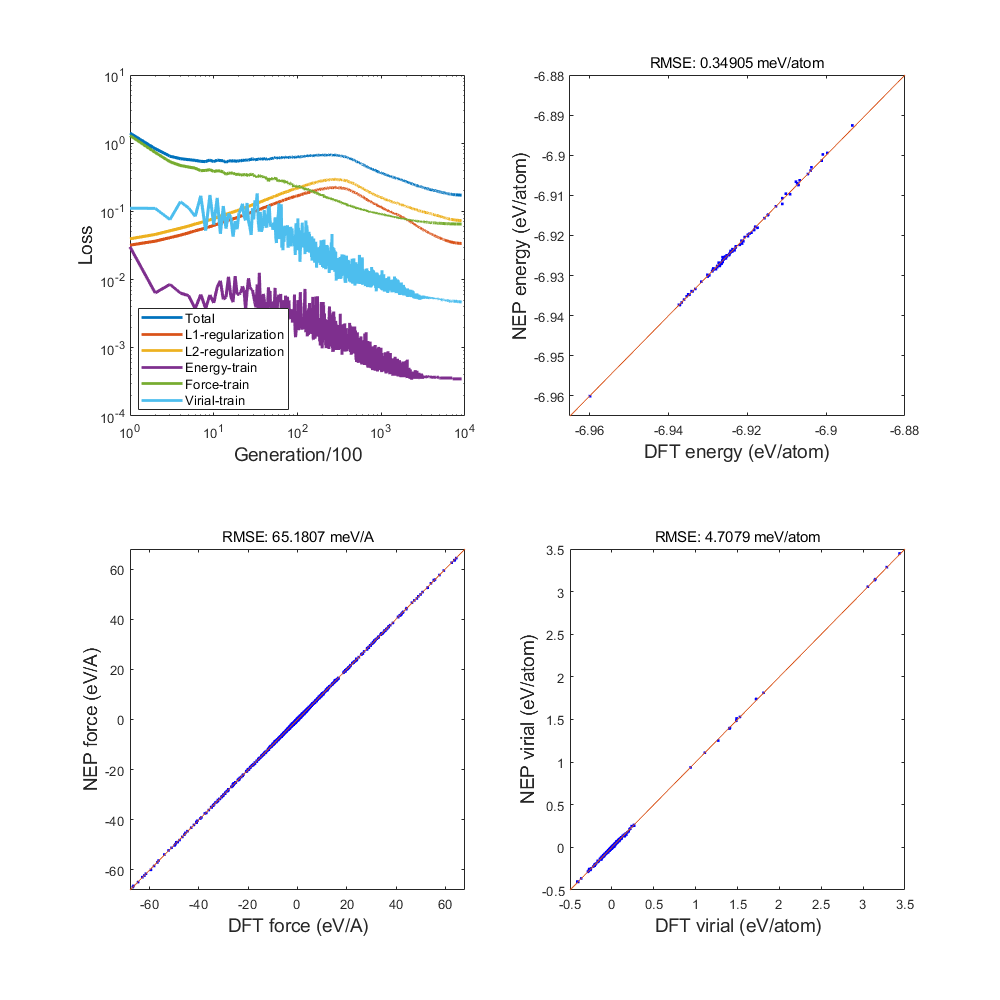
Fig.1 train loss
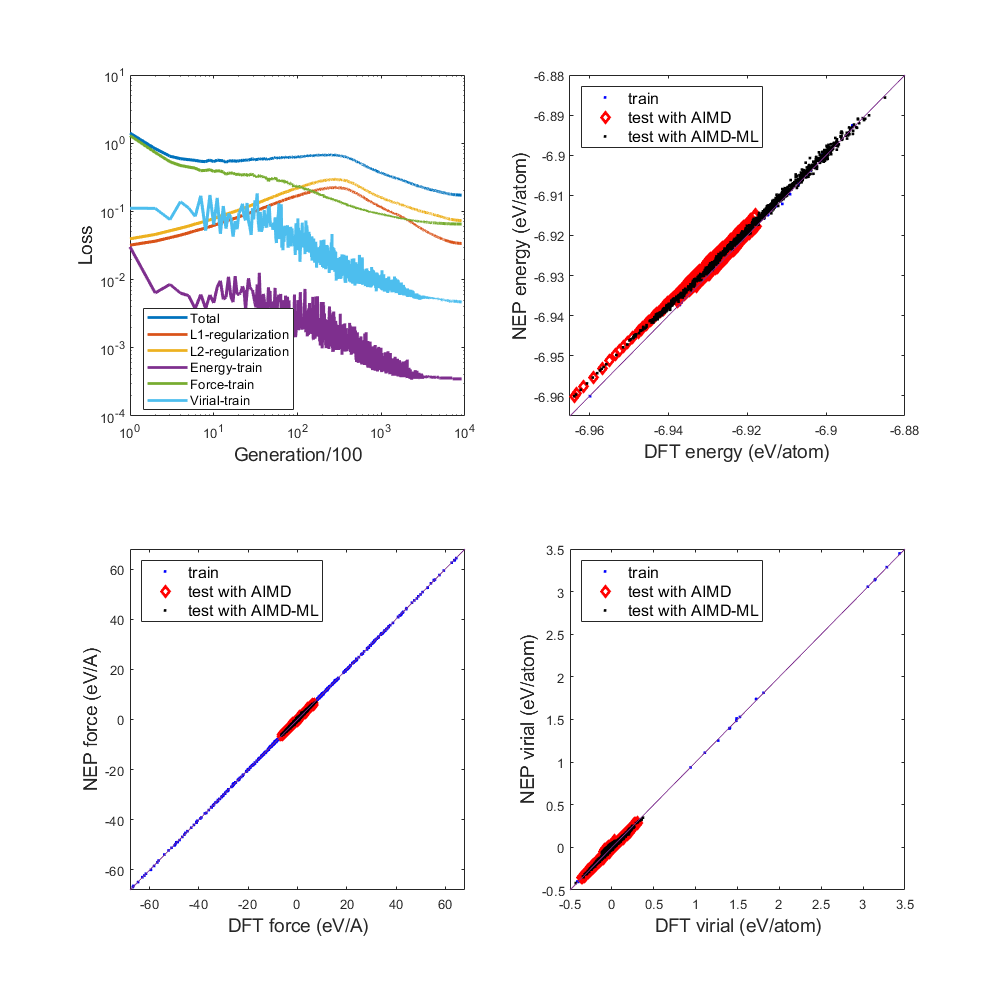
Fig.2 test loss
2) RDF of all atomic pairs
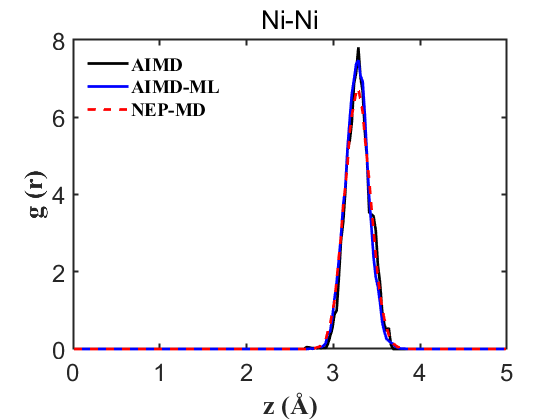
Fig.3 RDF Ni-Ni
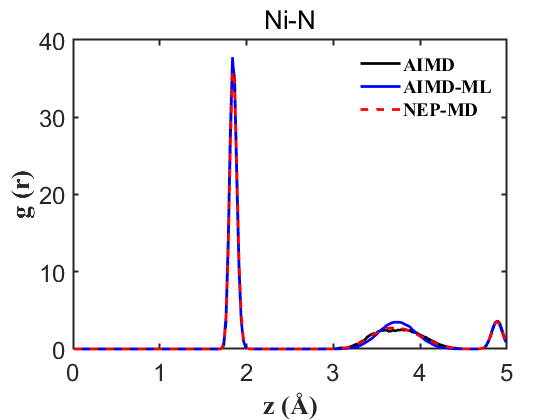
Fig.4 RDF Ni-N
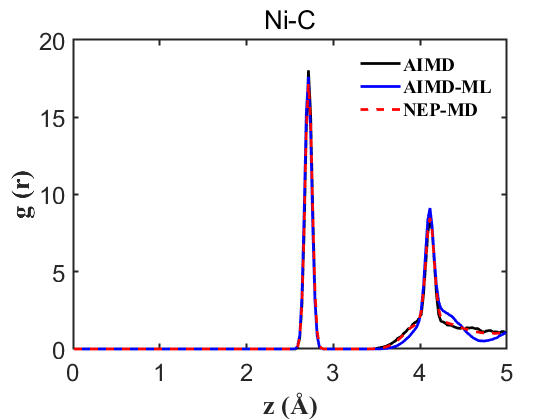
Fig.5 RDF Ni-C
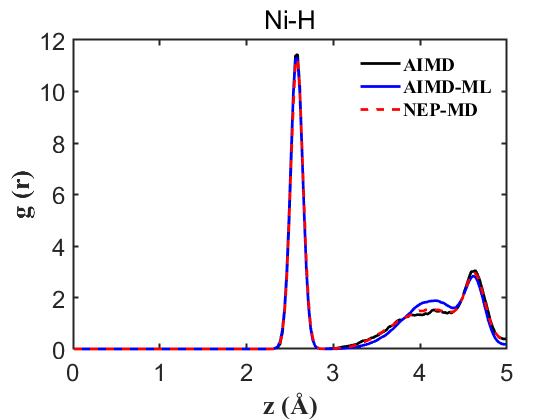
Fig.6 RDF Ni-H
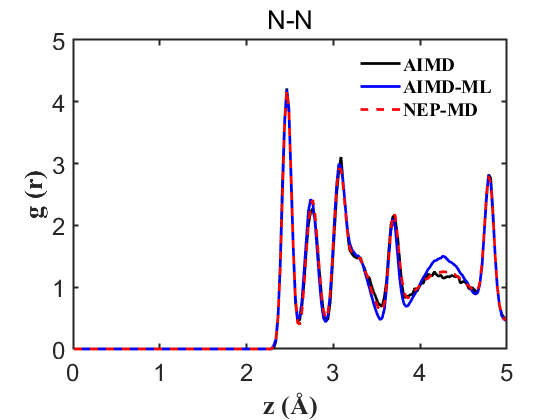
Fig.7 RDF N-N
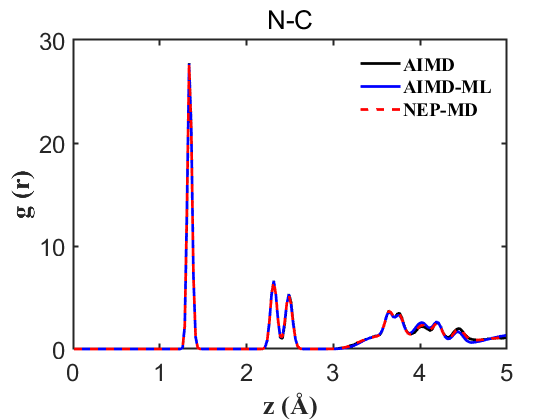
Fig.8 RDF N-C
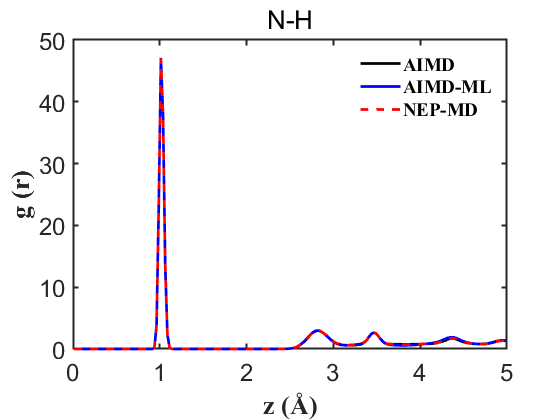
Fig.9 RDF N-H
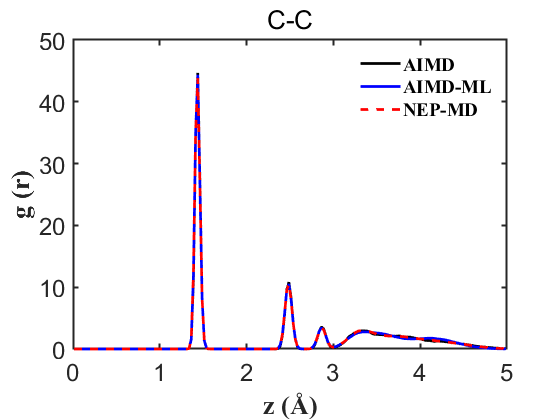
Fig.10 RDF C-C
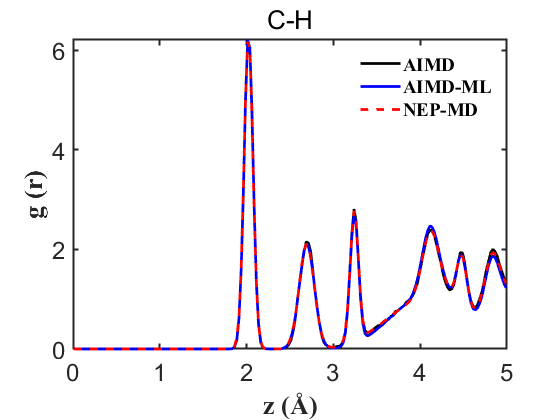
Fig.11 RDF C-H
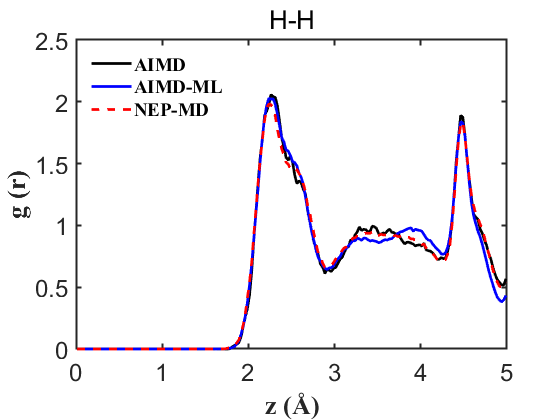
Fig.12 RDF H-H
3) Density
Tab.1 Density values calculated by three simulation methods
| Methods | Density (298K 1bar) |
|---|---|
| AIMD | 1.664 g/cm3 (05-10 ps) |
| AIMD_ML | 1.660 g/cm3 (25-50 ps) |
| NEP-MD | 1.664 g/cm3 (05-10 ns) |
Therefore, the above data strongly demonstrate the accuracy of our NEP even when only 107 structures are used. This greatly reduces our training cost and iteration time.
Appendix 1: Simulation set up
1) AIMD_ML_NPT
# basic parameters
SYSTEM = AIMDML_NPTcooling
#NCORE = 28 # 8*?=?
#KPAR = 4
#NPAR = 2
KGAMMA = .TRUE. # GAMMA point
KSPACING = 2.0 # to ensure that the k-mesh = 1*1*1, must test
ENCUT = 500.0 # to make the "Pullay stress" zero, a higher ENCUT is needed (>=600)
PREC = Normal # precision-mode
GGA = PE # GGA = PE
ISTART = 0 # read the WAVECAR file or not (ICHARG=2)
LWAVE = .FALSE. # write WAVECAR or not
LCHARG = .FALSE. # write CHGCAR or not
IVDW = 12
# Electronic Relaxation
ISMEAR = 0 # 0=Gaussian smearing. (1,2)=Methfessel-Paxton order N
SIGMA = 0.05 # width of the smearing in eV
EDIFF = 1E-5 # global break for electronic SC-loop
#EDIFFG = -1e-2 # break for force
LREAL = A # projection operators
NELM = 120 # maximum number of electronic SC
NELMIN = 5 # avoid breaking after 2 steps
# Molecular Dynamics
IBRION = 0 # Activate MD
MDALGO = 3 # 2=Nose-Hoover, 3=Langevin
ISIF = 3 # 1=NVE, 2=NVT, 3=NpT
#SMASS = 3
ALGO = Normal # Normal=IALGO=38 (Davidson), Fast=IALGO=48 (RMM-DIIS)
ISYM = 0 # no symmetry for MD, completely
TEBEG = 298 # Begin temperature K
TEEND = 298 # Final temperature K
NSW = 100000 # Max ionic steps
POTIM = 0.50 # Timestep in fs
LANGEVIN_GAMMA = 10.0 10.0 10.0 10.0 # damp (ps-1) for atom degrees-of-freedom
LANGEVIN_GAMMA_L = 10 # friction coefficient (ps-1) for lattice degrees-of-freedom
PMASS = 1000 # fictitious mass (in amu) to lattice degrees-of-freedom
PSTRESS = 1.01E-3 # controls the target pressure in AIMD
NWRITE = 1 # long MD-runs use NWRITE=0 or 1
NBLOCK = 1 # write PCF and DOS, scale kinetic energy, also the output interval of XDATCAR
# Machine learning force field
ML_LMLFF = .TRUE. # enables/disables the use of MLFF
ML_MODE = train # MLFF method (or mode)
ML_LBASIS_DISCARD = .FALSE.
ML_MB = 5000
2) Single point
Systerm=NiHAB
ENCUT=600
EDIFF=1E-6
ISMEAR=0
SIGMA=0.02
KSPACING=0.2
KGAMMA=.TRUE.
NELM=120
NELMIN=5
IVDW=12
ALGO=F
PREC=Normal
LREAL=Auto
GGA=PE
NSW=0
LWAVE=.FALSE.
LCHARG=.FALSE.
3) NEP Train
type 4 Ni N C H # this is a mandatory keyword
version 4 # default
cutoff 6 4 # default 8 4
n_max 8 6 # default 4 4
basis_size 12 12 # default
l_max 4 2 0 # default
neuron 30 # default
lambda_1 0.1 # default
lambda_2 0.1 # default
lambda_e 1.0 # default 0.01
lambda_f 1.0 # default 0.01
lambda_v 0.1 # default
batch 200 # default 1000
population 50 # default
generation 500000 # default 100000
4) LAMMPS NEP-MD
units metal #
boundary p p p#
atom_style atomic #
read_data data.start
pair_style nep ../nep.txt
pair_coeff * *
timestep 0.0005
thermo_style custom step time temp press density pe ke etotal #
thermo 100
dump trjnpt all custom 2000 mydump-npt.lammpstrj id type x y z
fix npt3 all npt temp 298 298 0.05 iso 1.0 1.0 0.5
run 20000000